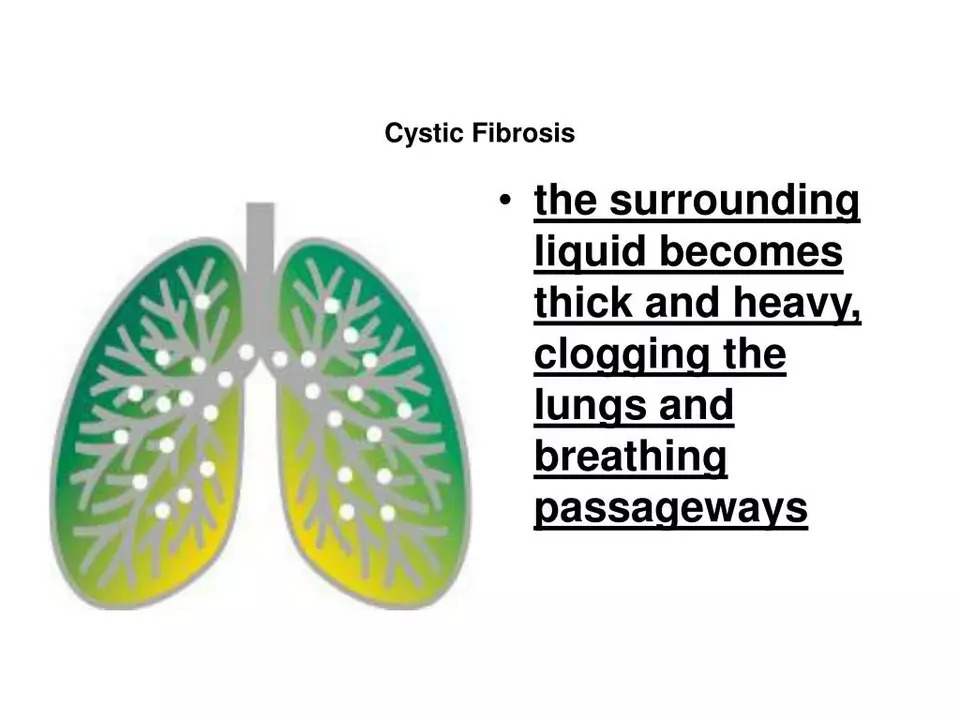
Cystic fibrosis (CF) is a genetic disease that makes mucus thick and sticky, clogging lungs and the digestive tract. People with CF face chronic lung infections, trouble gaining weight, and other organ problems. Thanks to better care and new medicines, many live into adulthood and lead active lives.
CF comes from changes in the CFTR gene passed from both parents; a simple blood test or newborn screening picks it up early. Genetic testing identifies the exact mutation and helps doctors choose the best medicines.
CFTR modulator drugs target the faulty protein in many people with CF. Names you might hear include ivacaftor, lumacaftor, tezacaftor, and elexacaftor — combinations of these can improve lung function and reduce flare-ups. Airway clearance matters: daily chest physiotherapy, devices like the vest, and exercise move mucus out of the lungs. Inhaled treatments — dornase alfa, hypertonic saline, and inhaled antibiotics such as tobramycin — thin mucus and fight infection. People with pancreatic involvement usually take enzymes with meals and follow a high-calorie diet.
Keep regular clinic visits with a CF center — they coordinate chest care, nutrition, and infection control. Know the signs of a flare: increased cough, thicker sputum, fever, or sudden weight loss. Antibiotics for lung infections may be oral, inhaled, or IV — follow the full course and ask about home IV options. Stay up to date with vaccines and avoid close contact with people who have colds or the flu. Good dental and sinus care help, too — these areas can harbor bacteria that reach the lungs.
If you or a family member carries CF, genetic counseling helps with planning and testing. Buying specialty drugs online can save money but be careful: use verified pharmacies and check that drugs come with proper labeling and a pharmacy contact. Never switch or stop CF medicines without talking to your CF team.
Clinical trials keep expanding options; ask your center about studies for new modulators or infection strategies. With routine care and modern drugs, many people with CF work, study, and travel — plan ahead for flights and medical supplies. Questions about treatments or where to find reliable info? Talk to your CF clinic or use trusted resources like national CF foundations.
Nutrition is a pillar of care: many people need high-calorie meals, vitamins A, D, E, K, and pancreatic enzymes to absorb fats. Regular weight checks and dietitian visits prevent malnutrition and support growth in kids. Physical activity helps clear mucus and build strength; small daily routines beat sporadic workouts. Mental health matters: CF care teams often include psychologists who help with stress, treatment burnout, and family concerns. Plan for emergencies: keep a list of current meds, allergies, hospital contacts, and clear instructions for ER staff. Know your infections: Pseudomonas and Staph aureus are common; your team will tailor antibiotics and airway care.
Newborn screening catches CF early; early treatment changes outcomes. Talk to a genetic counselor when planning a family. We're here to help you find reliable medication info.
I recently came across some interesting research about Amiloride, a drug that might have potential in treating Cystic Fibrosis. Cystic Fibrosis is a genetic disorder that mainly affects the respiratory and digestive systems, making it difficult for patients to breathe and digest food. Amiloride works by blocking certain channels in the cells, which could help reduce the thick mucus build-up commonly seen in Cystic Fibrosis patients. While this drug is not a cure, it could potentially improve the quality of life for those affected by the disorder. I'm excited to see how future studies on Amiloride will unfold and hope it can make a real difference for Cystic Fibrosis patients.
READ